Chemical Denaturation
In the less expensive technique of equilibrium unfolding, the fractions of folded and unfolded molecules (denoted as and, respectively) are measured as the solution conditions are gradually changed from those favoring the native state to those favoring the unfolded state, e.g., by adding a denaturant such as guanidinium hydrochloride or urea. (In equilibrium folding, the reverse process is carried out.) Given that the fractions must sum to one and their ratio must be given by the Boltzmann factor, we have
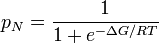

Protein stabilities are typically found to vary linearly with the denaturant concentration. A number of models have been proposed to explain this observation prominent among them being the denaturant binding model, solvent-exchange model (both by John Schellman) and the Linear Energy Model (LEM; by Nick Pace). All of the models assume that only two thermodynamic states are populated/de-populated upon denaturation. They could be extended to interpret more complicated reaction schemes.
The denaturant binding model assumes that there are specific but independent sites on the protein molecule (folded or unfolded) to which the denaturant binds with an effective (average) binding constant k. The equilibrium shifts towards the unfolded state at high denaturant concentrations as it has more binding sites for the denaturant relative to the folded state . In other words, the increased number of potential sites exposed in the unfolded state is seen as the reason for denaturation transitions. An elementary treatment results in the following functional form:
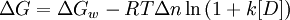
where is the stability of the protein in water and is the denaturant concentration. Thus the analysis of denaturation data with this model requires 7 parameters: ,, k, and the slopes and intercepts of the folded and unfolded state baselines.
The solvent exchange model (also called the ‘weak binding model’ or ‘selective solvation’) of Schellman invokes the idea of an equilibrium between the water molecules bound to independent sites on protein and the denaturant molecules in solution. It has the form:

where is the equilibrium constant for the exchange reaction and is the mole-fraction of the denaturant in solution. This model tries to answer the question of whether the denaturant molecules actually bind to the protein or they seem to be bound just because denaturants occupy about 20-30% of the total solution volume at high concentrations used in experiments, i.e. non-specific effects – and hence the term ‘weak binding’. As in the denaturant-binding model, fitting to this model also requires 7 parameters. One common theme obtained from both these models is that the binding constants (in the molar scale) for urea and guanidinium hydrochloride are small: ~ 0.2 for urea and 0.6 for GuHCl.
Intuitively, the difference in the number of binding sites between the folded and unfolded states is directly proportional to the differences in the accessible surface area. This forms the basis for the LEM which assumes a simple linear dependence of stability on the denaturant concentration. The resulting slope of the plot of stability versus the denaturant concentration is called the m-value. In pure mathematical terms, m-value is the derivative of the change in stabilization free energy upon the addition of denaturant. However, a strong correlation between the accessible surface area (ASA) exposed upon unfolding, i.e. difference in the ASA between the unfolded and folded state of the studied protein (dASA), and the m-value has been documented by Pace and co-workers. In view of this observation, the m-values are typically interpreted as being proportional to the dASA. There is no physical basis for the LEM and it is purely empirical, though it is widely used in interpreting solvent-denaturation data. It has the general form:

where the slope is called the "m-value"(> 0 for the above definition) and (also called Cm) represents the denaturant concentration at which 50% of the molecules are folded (the denaturation midpoint of the transition, where ).
In practice, the observed experimental data at different denaturant concentrations are fit to a two-state model with this functional form for, together with linear baselines for the folded and unfolded states. The and are two fitting parameters, along with four others for the linear baselines (slope and intercept for each line); in some cases, the slopes are assumed to be zero, giving four fitting parameters in total. The conformational stability can be calculated for any denaturant concentration (including the stability at zero denaturant) from the fitted parameters and . When combined with kinetic data on folding, the m-value can be used to roughly estimate the amount of buried hydrophobic surface in the folding transition state.
Read more about this topic: Equilibrium Unfolding
Famous quotes containing the word chemical:
“We are close to dead. There are faces and bodies like gorged maggots on the dance floor, on the highway, in the city, in the stadium; they are a host of chemical machines who swallow the product of chemical factories, aspirin, preservatives, stimulant, relaxant, and breathe out their chemical wastes into a polluted air. The sense of a long last night over civilization is back again.”
—Norman Mailer (b. 1923)